|
Cuando se habla de ADN antiguo (ADNa)
parece inevitable hacer una referencia a "Parque Jurásico",
película basada en el hecho de que un grupo de científicos
logran traer a la vida a diferentes especies de dinosaurios,
a partir del ADN fósil recuperado en insectos preservados
en ámbar.
Aunque, ha sido posible recuperar
pequeños fragmentos de ADNa a partir de los restos fosilizados
de un dinosaurio de 80 m. a. (Woodward et al., 1994), actualmente
la ficción supera aún a la realidad. No disponemos
de la tecnología necesaria para llevar a cabo un proyecto
tan ambicioso como "Parque Jurásico", puesto
que, la degradación existente en la estructura molecular
del ADNa limita en gran medida las posibilidades de análisis,
además de otras limitaciones aún de mayor importancia.
Normalmente el tamaño del fragmento de ADN secuenciado
no supera los 300 pares de bases (pb) de longitud, mientras que
el genoma completo de un organismo puede llegar a presentar aproximadamente
3.000 millones de pb.
Sin embargo si es posible recuperar
y analizar pequeños fragmentos de ADN fósil (o
ADN antiguo, ADNa). Uno de los trabajos pioneros en este campo
fue la clonación y secuenciación de un fragmento
de ADN de 220 pb de longitud a partir del tejido taxidermizado
de un animal extinguido hace 150 años (Equus quagga)
(Higuchi et al., 1984). Uno de los principales problemas con
los que estos autores se encontraron fue una baja eficiencia
de clonación, fundamentalmente debido a la mala preservación
del ADNa.
Estos primeros trabajos con ADNa
pudieron haberse quedado como una mera curiosidad, si no hubiera
sido por el desarrollo de una nueva metodología de análisis
molecular, la Reacción en Cadena de la Polimerasa
(PCR) (Mullis & Faloona, 1987). Esta técnica implica
la síntesis en el laboratorio de millones de copias de
un fragmento de ADN a partir de una pequeña cantidad inicial
(Saiki et al., 1985; Mullis & Faloona, 1987). Es un proceso
cíclico en el que la cantidad de una determinada secuencia
de ADN elegida por nosotros, se incrementa exponencialmente hasta
niveles que permitan la aplicación de las diferentes técnicas
disponibles para su análisis (secuenciación y polimorfismos
de restricción). La PCR requiere el diseño de "primers"
o "cebadores" específicos, que son secuencias
de ADN de 18-25 pb de longitud, complementarias a la molécula
de ADN de interes (ADN molde), y que determinan el principio
y el final de la región que se quiere amplificar. A partir
de estos "cebadores", un enzima denominado Taq
polimerasa irá copiando la molécula de ADN molde.
Obviamente una de las aplicaciones
más interesantes de esta nueva metodología, es
la posibilidad de estudiar de manera directa la composición
genética de las poblaciones del pasado, compararla con
las del presente, e inferir los procesos evolutivos que han tenido
lugar a lo largo de la historia. Si bien la genética de
poblaciones ha desarrollado sofisticados métodos estadísticos
para inferir las relaciones filogenéticas y la historia
evolutiva de las poblaciones, en particular de las poblaciones
humanas, el análisis del ADNa permite construir hipótesis
basadas no en inferencias estadísticas sino en datos directos.
La perspectiva espacio-temporal que esta metodología proporciona
permitirá ofrecer respuestas cada vez más precisas
a las viejas cuestiones de ¿quiénes somos? y ¿de
dónde venimos?
En este contexto, el objetivo
principal planteado en este trabajo ha sido obtener datos genéticos
directos de un conjunto de poblaciones de procedencia arqueológica,
distribuidas por el territorio vasco y realizar la comparación
con las poblaciones actuales. Asi, hemos analizado 121 muestras
recuperadas en los yacimientos de: SJAPL (Araba): 5070±150,
5020±140 BP (Etxeberria & Vegas, 1988), Longar (Nafarroa):
4445±70, 4580±90 BP (Armendariz & Irigarai,
1995) y Pico Ramos (Bizkaia): 4100±110, 4790±110
BP (Zapata, 1995).
Problemas Metodológicos
y Optimización de la Metodología
Cuando se trabaja con
ADNa, resulta primordial establecer unas pautas o criterios de
esterilidad que garanticen la autenticidad de los resultados
obtenidos, debido al alto riesgo de contaminación con
ADN moderno y a la escasez de moléculas de ADN endógeno
del tejido antiguo (hueso o diente). Asi, es imprecindible la
eliminación y posterior esterilización de las superficies
externas de los tejidos, la extracción del ADN en un laboratorio
exclusivamente dedicado al trabajo con ADNa, la separación
física de los procesos pre-PCR (extracción y preparación
de la reacción de amplificación) y post-PCR (tipaje
de las muestras amplificadas), la esterilización de reactivos
y aparataje mediante luz UV, la realización de controles
de contaminación en los diferentes pasos del procesamiento
de la muestra, la realización de múltiples extracciones
del mismo individuo y el análisis en dos laboratorios
independientes, a fin de corroborar los resultados obtenidos,
y finalmente el tipaje obtenido debe mostrar sentido filogenético
(Stoneking, 1995).
Las piezas dentarias han resultado
el resto biológico de mayor utilidad en cuanto a rendimiento
de ADN útil, en comparación con el tejido óseos
(hueso). Tras un intenso proceso de decontaminación, basado
en la limpieza de las superficies dentarias mediante una mezcla
de ácidos, el ADN se extrae de la pulpa dentaria siguiendo
el método del fenol-cloroformo (Hagelberg & Clegg,
1991). El estado fragmentado del ADNa conlleva que normalmente
se recuperen fragmentos menores de 200 nucleótidos de
longitud.
La mayoría de los trabajos
con ADNa se han centrado en el análisis del ADN mitocondrial
(ADN mt), ya que cada célula (eucariota) presenta entre
3.000-5.000 copias de genomas mitocondriales (Shuster et al.,
1988), lo cual facilita enormemente su supervivencia y recuperación,
en comparación con el ADN nuclear. Además, otras
ventajas que presenta el estudio del ADNmt son que el genoma
mitocondrial humano moderno está secuenciado en su totalidad
y sus polimorfismos están estudiados en un gran número
de poblaciones; asimismo la rápida tasa de evolución
y su herencia materna, hacen de él un marcador genético
útil para establecer afinidades poblacionales entre grupos
humanos.
El análisis del ADNmt
se puede abordar desde dos perspectivas:
1) secuenciación y 2) enzimas de restricción.
La secuenciación implica
la "lectura" de una cadena de nucleótidos. La
comparación de las secuencias provenientes de distintos
individuos, permite averiguar en cuantas posiciones difieren
dos o más muestras de ADN y establecer asi relaciones
filogenéticas o de parentesco evolutivo entre ellas. Para
analizar secuencias de unos 400 pb, es necesario la amplificación
y secuenciación independiente de varios fragmentos solapantes
de unos 100 pb de longitud, hasta completar la región
deseada. Por otro lado, algunos nucleótidos suelen estar
modificados en las moléculas de ADNa debido a reacciones
químicas de oxidación e hidrólisis, por
lo que durante la amplificación enzimática, la
Taq polimerasa puede introducir nucleótidos erróneos
en estos lugares modificados. Todo ello conlleva que cada fragmento
se deba secuenciar varias veces para validar la información
obtenida.
El otro tipo de análisis,
los polimorfismos detectados mediante enzimas de restricción,
no se ve dificultado por estos errores tan marcadamente. Los
enzimas de restricción son moléculas capaces de
cortar (o digerir) el ADN en posiciones en las que se encuentre
presente determinadas secuencias diana (de 4 a 6 pb de longitud,
diferentes en general para cada enzima de restricción).
Los diferentes patrones de presencia/ausencia de corte para las
diferentes secuencias diana analizadas permite establecer de
manera alternativa a la secuenciación, las relaciones
genéticas entre las distintas muestras. Metodológicamente,
esta técnica es más sencilla y rápida, lo
que permite, por un lado la realización de un mayor número
de análisis en menos tiempo y por otro, mayor probabilidad
de obtener resultados positivos
El análisis de 7 polimorfismos
mediante la técnica de los enzimas de restricción
(Richards et al., 1998; Macaulay et al., 1999; Torroni et al.,
1998), permite clasificar la variabilidad del ADNmt de caucasoides
en 9 haplogrupos. Para ello, mediante la técnica de la
PCR se amplifican en cada muestra 7 fragmentos de unos 100 pb
de longitud, cada uno comprendiendo su respectiva secuencia diana
de interés. Tras la amplificación de estos fragmentos,
se verifica, mediante el análisis de los diferentes controles,
que no se ha producido contaminación durante el procesamiento
de la muestra. Si es así, cada fragmento se someterá
a digestión con su correspondiente enzima. Dependiendo
del modelo de digestión que presenta una muestra en cada
uno de los 7 nucleótidos analizados, se clasifica como
perteneciente a alguno de los 9 haplogrupos mitocondriales descritos
en caucasoides (Torroni et al., 1996).
Resultados Obtenidos en el
Análisis Genético de los Restos Prehistóricos
En nuestro estudio de
ADNmt prehistórico mediante la técnica de enzimas
de restricción, se han podido tipar con éxito el
97% de las muestras que presentaban todos los controles de la
contaminación negativos. Aunque esta cifra pueda parecer
elevada, hay que destacar que tan sólo se han analizado
el 25% de las muestras recuperadas, habiéndose seleccionado
exclusivamente las piezas dentarias intactas, para minimizar
la posibilidad de que algún tipo de ADN exógeno
contaminante (moderno o bacteriano) se infiltrara al interior
de la pulpa dental.
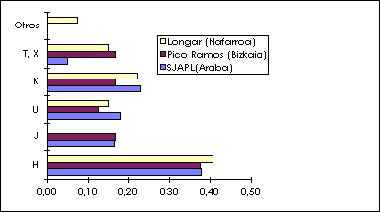
En la Fig.
1, se presentan los primeros datos genéticos obtenidos
en población prehistórica del País Vasco.
La comparación de las muestras prehistóricas, indica
que la población de Longar presenta la mayor diferenciación
genética en relación a las demás, debido
a la ausencia de individuos pertenecientes al haplogrupo J y presencia
de individuos que no se pueden clasificar en ninguno de los 9
haplogrupos descritos actualmente en caucasoides. También
desde el punto de vista arqueológico, el hipogeo de Longar
presenta unas características diferenciadas de otras estructuras
funerarias cercanas, más o menos contemporáneas,
como son los dólmenes de la Rioja Alavesa (Armendariz &
Irigarai, 1995). Por otro lado, el análisis de caracteres
no-métricos del cráneo(Izagirre et al.,
en prensa), indica semejanzas entre SJAPL (Araba) y Longar (Nafarroa).
Con los datos disponibles actualmente resulta difícil reconciliar
ambos tipos de evidencia (morfológica y genética)
en estas poblaciones prehistóricas. Un factor importante
a tener en cuenta en el análisis de los datos del ADNmt,
es que éstos nos indican la historia de los linajes femeninos,
la cual se verá afectada por una posible migración
diferencial entre ambos sexos (Seielstad et al., 1998). La existencia
de datos genéticos y craneales de un mayor número
de yacimientos prehistóricos podría ayudar a resolver
esta cuestión.
Asimismo, la comparación
de los resultados obtenidos en las poblaciones prehistóricas,
con los datos de otras poblaciones de vascos actuales, nos permiten
discutir una reciente hipótesis propuesta en base a datos
genéticos de la población actual del País
Vasco(Torroni et al. 1998), donde se considera que el haplogrupo
V, dada su alta frecuencia en esta región, constituye
un marcador de una expansión poblacional ocurrida en el
Paleolítico Superior, desde el sudoeste de Europa (País
Vasco) hace 10.000-15.000 años BP.
Los resultados obtenidos, ponen de manifiesto
la ausencia del haplogrupo V en las poblaciones prehistóricas
analizadas en este trabajo (Fig. 1).
Esta discrepancia en la frecuencia del haplogrupo V entre los
grupos actuales y prehistóricos del País Vasco,
puede tener diversas explicaciones:
|
1) La existencia de heterogeneidad
genética en la población del País Vasco.
Si la heterogeneidad que observamos actualmente (Aguirre et al.,
1991; Calderón et al. 1993; Manzano et al., 1996; Iriondo
et al., 1999), ocurrió también en la prehistoria,
la deriva genética debió ser la responsable de
que el haplogrupo V alcanzara una frecuencia elevada en una o
varias subpoblaciones del País Vasco y estuviera ausente
en otras. |
|
2) El tiempo de origen del haplogrupo
V puede ser más reciente que el propuesto (~10.000-15.000
B.P.) (Torroni et al. 1998), lo que explicaría su ausencia
en las muestras prehistóricas estudiadas en el presente
trabajo. Estas dataciones basadas en la variabilidad del ADNmt
actual son función de parámetros (tiempo de divergencia),
para los cuales diferentes autores ofrecen estimas que difieren
substancialmente. Los valores que se utilizan para la región
no codificante del ADNmt, oscilan entre 7% y 22%/millón
año (Tamura & Nei, 1993; Horai et al., 1995), lo que
conduce a dataciones muy variables. |
|
3) Que el haplogrupo V fuera introducido
en el País Vasco por un proceso de inmigración.
Una reciente hipótesis propone que un pequeño grupo
neolítico procedente del Caúcaso, habría
reemplazado a los cazadores-recolectores existentes en el País
Vasco hace ~5.000-5.500 B.P. (Calderón et al., 1998).
Sin embargo, este movimiento poblacional no explicaría
la discrepancia de datos que observamos, ya que la población
actual del Caúcaso presenta una frecuencia del 0% para
el haplogrupo V (Torroni et al., 1998). |
|
4) La interpretación del
registro arqueológico. La "expansión poblacional
ocurrida a finales del Paleolítico desde el sudoeste al
noreste de Europa" (Torroni et al., 1998), se ha fundamentado
en interpretaciones arqueológicas tipologistas, que explican
el parecido en el arte e industrias magdalenienses (10.000 B.P)
en el sudoeste de Francia y otras regiones europeas (Bélgica,
el Rhin, Suiza, Moravia y Polonia), en términos de migración/difusión
(Otte, 1990). Sin embargo, este tipo de interpretaciones es muy
criticada (Clark & Lindly, 1991), y resulta más aceptable,
otro tipo de enfoques enmarcados en un ámbito ecológico.
La expansión de las áreas de ocupación en
Europa después del último periodo glacial (~12.000-10.000
B.P.), se dió en dirección sur-norte, ya que la
mejoría climática produjo cambios en el paísaje
y modos de subsistencia, que favorecieron la ocupación
de áreas (en norte y medio Europa), que hasta entonces
habían estado escasamente habitadas por grupos de cazadores
dispersos en amplios territorios (Straus, 1996). |
Basándonos en todo lo argumentado
anteriormente, proponemos que la mutación que define el
haplogrupo denominado V, pudo haber alcanzado valores muy elevados
en algunas regiones del País Vasco (Gipúzkoa, 20%),
por efecto de la deriva genética, lo que explicaría
la heterogeneidad observada para este haplogrupo entre distintas
muestras (entre 0 y 20%) (Izagirre & de la Rúa, 1999).
Ésto debió ocurrir en el periodo pre-mesolítico
(> 11.000 B.P.) cuando el tamaño efectivo de los grupos
humanos era suficientemente pequeño como para que la deriva
genética tuviera un efecto apreciable, ya que a partir
de entonces se dió un cambio demográfico sustancial
en el País Vasco (de la Rúa, 1995). Aunque esta
explicación es compatible con la datación atribuída
al origen de la mutación, no es preciso recurrir a modelos
migracionistas para explicar la distribución del haplogrupo
V en el resto de las poblaciones indoeuropeas.
El análisis molecular
de restos arqueológicos, a pesar de las limitaciones inherentes
al tamaño muestral recuperado y al número y tipo
de marcadores genético-poblacionales que se pueden analizar,
resulta útil, tanto para valorar hipótesis planteadas
en base a los datos genéticos de la población actual,
como para el establecimiento de hipótesis basadas en datos
genéticos directos de las poblaciones prehistóricas.
Sin embargo, es necesario disponer de un mayor número
de muestras poblacionales que comprendan la distribución
de la variabilidad actual y pasada, para poder reconstruir con
mayor solidez las relaciones filogenéticas existentes
entre los grupos humanos del País Vasco. Este trabajo
representa un primer paso en esta dirección.
Figura 1. Distribución de la frecuencia
de los haplogrupos del ADNmt en las poblaciones prehistóricas
de SJAPL (Araba), Pico Ramos (Bizkaia) y Longar (Nafarroa).
(VOLVER)
Agradecimientos:
Este trabajo se ha realizado gracias a las ayudas concedidas
al proyecto titulado: "Aportación de la biología
molecular al estudio de la evolución biológica
de la población del País Vasco" por la Diputación
Foral de Bizkaia, Diputación Foral de Alava, Fundación
Caja Vital y BBK, así como al proyecto GN 154.310-0001-94
(Gobierno de Navarra y U.P.V./E.H.U.). |
|
Aguirre A et al. (1991). Am. J. Hum. Genet., 49: 450-458
Armendariz J & Irigarai S (1995). Centro de Estudios
Tierra-Estella, Lizarraldeko Ikastetxea
Calderón R et al. (1992). Ann. Hum. Biol., 20:
109-120
Calderón R. et al. (1998). Hum. Biol., 70: 667-698
Clark GA & Lindly JM (1991). Curr. Anthropol. 32:
577-587
de la Rúa C. (1995). El passat dels Pirineus des
de'una perspectiva multidisciplinaria. Muntanyes I Poblacio.
Etxeberria F & Vegas JI (1988). Munibe, 6: 105-112
Hagelberg E & Clegg JB (1991). Procc. R. Soc. Lond.
B, 244: 45-50
Higuchi RG et al. (1984). Nature, 312: 282-284
Horai S et al. (1995). Procc. Natl. Acad. Sci USA, 92:
532-536
Iriondo M et al. (1999). Rev. Esp. Antrop. Biol., 20:
181-193
Izagirre N & de la Rúa C (1999). Am. J. Hum.
Genet., 65: 199-207
Izagirre N et al. (en prensa). Molecular genetics and
early Europe: working papers in population prehistory. The McDonald
Institute, Cambridge
Macaulay V et al. (1999). Am. J. Hum. Genet., 64: 232-249
Manzano C et al. (1996). Am. J. Phys. Anthropol., 99:
249-258
Mullis KB & Faloona FA (1987). Meth. Enzymol, 155:
335-350
Otte M (1990). The world at 18,000 BP. Vol. 1. London,
pp. 54-68
Richards MB et al. (1998). Ann. Hum. Genet., 62: 241-260
Saiki RK et al. (1985). Science, 230: 1850-1854
Seielstad MT et al. (1998). Nature Genetics, 20: 278-280
Shuster RC et al. (1988). Biochem. Biophys. Res. Commun.,
155: 1360-1365
Stoneking M (1995). Am. J. Hum. Genet., 57: 1259-1262
Strauss L (1996). Humans at the end of the Ice Age. New
York
Tamura K & Nei M (1993). Mol. Biol. Evol., 10: 512-526
Torroni A et al. (1996). Genetics, 144: 1835-1850
Torroni A et al. (1998). Am. J. Hum. Genet., 62: 1137-1152
Woodward SR et al (1994). Science, 266: 1229-1232
Zapata L (1995). Munibe, 68: 185-199 |
N. Izagirre &
C. de la Rúa, Euskal Herriko Unibertsitatea. Zientzi
Fakultatea. Animali Biologia eta Genetika Saila. Antropologiako
Laborategia. Posta kutxa, 644. 48080 Bilbo |

